Summary
The Q-HOP method is a semi-quantitative approach to study proton transport in biomolecular systems. It can efficiently explore proton transfer paths e.g., in proteins that consisting of many subsequent transfer events.
Researcher
Wei Gu
Publication
Lill, M.A. and Helms, V., (2001) Journal of Chemical Physics, vol. 115, p. 7993-8005, doi: 10.1063/1.1407293. Molecular dynamics simulation of proton transport with quantum mechanically derived proton hopping rates (Q-HOP MD)
Description
Classical molecular dynamics simulations employ empirical molecular mechanics force fields where chemical bonds are modelled as harmonic springs. Therefore, chemical bonds defined in the molecular topology file at the start of a simulation remain bonded throughout the simulation thus prehibiting the modelling of any chemical reactions. Theoretical studies of chemical reactions are, however, routinely performed nowadays using (CPU intensive) quantum chemical techniques. In the field of enzymatic reactions, it is common to couple an inner quantum region including the “active site” where chemical reactions take place to an outer region modelled by molecular mechanics. This separation into two regions remains constant during a dynamics simulation. These techniques are, therefore, not suited to treat proton transport through entire membrane proteins that would require a travelling quantum region. A very efficient simulation scheme was developed by the Helms group over the past 4 years where instantaneous proton hopping events are included in MD simulations of arbitrary biomolecular or organic systems. The hopping rates are carefully parameterized against accurate quantum chemical calculations (Lill00, Lill01a, Lill01b, Lill01c, Herzog 06). The first application to a protein system was the proton shuttle in green fluorescent protein (Lill02).
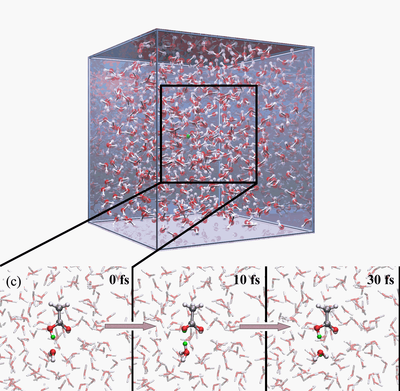
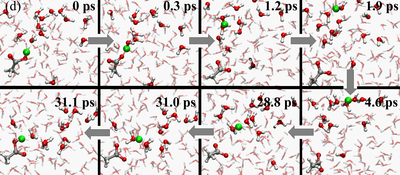
The Q-HOP MD is now being implementated into the parallel quantum chemistry package NWChem. In addtion, we have optimized a separate set of atomic point charges on the donor and acceptor groups to mimic the environmental energy differences using the DFT/B3LYP theory. Base on these progress, we have generated 50-ns Q-HOP MD trajectories of solvated acetic acid. Without imposing any particular transfer mechanisms, we characterized the protonation equilibria from the observed spontaneous partitioning between titratable sites and solvent molecules (see figures above). During the simulation, two different regimes of proton transfer were observed. Extended phases of frequent proton swapping between acetic acid and nearby water were separated by phases where the proton freely diffuses in the simulation box until it is captured again by acetic acid (see figures above). The pKa of acetic acid was calculated around 3.0 based on the relative population of protonated and deprotonated states and the diffusion coefficient of excess proton was computed from the average mean squared displacement in the simulation. Both calculated values agree well with the experimental measurements.