Summary
QVADIS is a general Java3D program package to characterize proton transfer pathways in biomolecules accounting for side chain flexibility and transfer kinetics.
Researcher
Kerstin Kunz
Publication
Kunz, K. and Helms, V. (2007) GI-Edition – Lecture Notes in Informatics (LNI) – Proceedings, vol. 115. QVADIS: A Package to Compute Proton Transfer Pathways in Proteins. (Eds.) Falter, C., Schliep, A., Selbig, J., Vingron, M., and Walther, D.
Description
Reactions involving proton transfer are crucial in many areas of biology such as in enzyme catalysis and in bioenergetics, but also in other areas of chemistry and material science such as in hydrogen fuel cells. Our own interest is the identification of proton pathways through integral membrane pumps that are the basis for establishing a membrane potential and the synthesis of ATP.
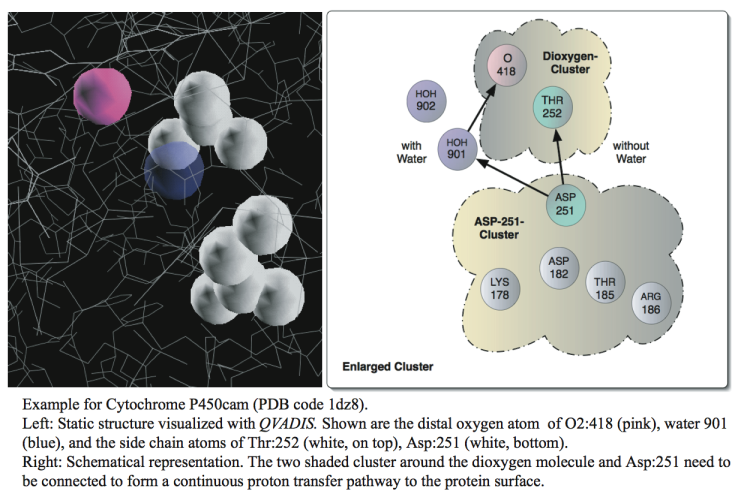
As experimental techniques are facing fundamental difficulties in resolving these long-range pathways in biomolecules, there is great promise for developing computational methods that may step in. Establishing these pathways may guide further experimental studies or the design of proton pumps with tailored conduction properties.
Recent experimental and computational work showed that proton conduction in proteins mostly proceeds along hydrogen-bonded chains involving amino acid side chains and buried water molecules. The protons migrate as structural defects via the well-known Grotthuss mechanism [Mar06]. Importantly, the proton(s) will distribute over all kinetically accessible titratable groups of the protein subject to their local pKa values [SW04].
The dynamics of proton transfer is determined by the energy barriers between different states, often involving reorientations of side chains and water molecules. Therefore, proton conduction is intrinsically a dynamic phenomenon that is not well understood on the basis of static crystal structures of proteins. In 2003, Taraphder et al. [TH03a][TH03b] presented an algorithm to characterize putative proton transfer pathways in proteins along hydrogen bonded networks. This algorithm accounts for side chain flexibility and scores the detected pathways on the basis of a simple steric energy function.
Unfortunately, their package is not publicly available. On the other hand, we have previously developed a parametrized scheme termed Q-HOP to compute proton transfer probabilities per time interval during classical molecular dynamics simulations [LH01a] [LH01b]. Input parameters are the chemical nature of donor and acceptor atom as well as their distance. In 2006, Herzog et al. [HFHL06] extended the parameter set to transfers between all titratable amino acids and involving water. This parametrization allowed us for the first time to simulate the proton equilibrium between acetic acid and a water box from unbiased molecular dynamics simulations [GFSH07] in good agreement with the experimental pKa value. By combining Q-HOP with the Taraphder-Hummer algorithm, we present here for the first time a general program package to characterize proton transfer pathways in biomolecules accounting for side chain flexibility and transfer kinetics that we implemented in Java3D.
While Java3D is compatible with different platforms and allows performing remote applications, it has certain deficits related to its performance concerning interaction speed or its ability to load large molecules. In 2003, Can et al. [CWWS03] introduced a method to tackle these problems. Their FPV method was used here as skeletal structure for our scene graph concept to share the main functionalities of loading and reading a PDB file and creating a static scene in which required objects can be inserted.
Our current model is based on several assumptions. First of all, we assume that the proton can freely partition within each static cluster due to the local pKa values of the amino acids. This leads to an enormous simplification of the search problem as the only degrees of freedom to be searched are the side chain dihedral angles of those residues at the cluster boundaries that were previously found in a coarse scan as putative candidates to enlarge the cluster. In this step, the protein backbone is kept fixed and only side chain rotations of the two contacting residues are exhaustively sampled. This latter approximation appears quite reasonable. The mobility of the buried water molecules was modeled implicitly by reducing their distance from potential donor and acceptor atoms by a given distance value.
In future work, as quite large energy barriers can be encountered due to collisions of the rotated side chains with neighboring groups, we may find it advantageous to allow for simultaneous relaxation of those colliding side chains. Also, we plan to combine this approach with electrostatic continuum calculation of local pKa values [KBU06] [BBU06] which will provide a baseline for the relative population of local clusters. When estimating the proton transfer energetics, a future version of QVADIS will also include the electrostatic interaction with the environment.
In summary, we have introduced a novel software package QVADIS to discover proton transfer pathways in biomolecules and to score their transfer kinetics by combining a variant of a previously published algorithm by Taraphder & Hummer and of our Q-HOP methodology. As proton transfer reactions are crucial in many areas of biology and material sciences, this tool may assist researchers in obtaining deeper understanding of the transfer processes and, eventually, to design macromolecules with desired transfer characteristics.
References
- [BBU06] E. Bombarda, T. Becker, and G. M. Ullmann. The Influence of the Membrane Potential on the Protonation of Bacteriorhodopsin: Insights from Electrostatic Calculations into the Regulation of Proton Pumping. J. Am. Chem. Soc., 128:12129–12139, 2006.
- [BWF00] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, and P.E. Bourne. The Protein Data Bank. Nucl. Ac. Res., 28:235–242, 2000.
- [CWWS03] T. Can, Y. Wang, Y.F. Wang, and J. Su. FPV: Fast protein visualisation using java 3d. Bioinformatics, 19(8):913–922, 2003.
- [GFSH07] W. Gu, T. Frigato, T.P. Straatsma, and V. Helms. Dynamisches Protonengleichgewicht der in Wasser gelösten Essigsäure. Angewandte Chemie, 2007.
- [HFHL06] E. Herzog, T. Frigato, V. Helms, and C.R.D. Lancaster. Energy barriers of proton transfer reactions between amino acid side chain analogs and water from ab initio calculations. J. Comput. Chem., 27:1534–1547, 2006.
- [KBU06] A. R. Klingen, E. Bombarda, and G. M. Ullmann. Theoretical investigation of the behavior of titratable groups in proteins. Photochem. Photobiol. Sci, 5:588–596, 2006.
- [LH01a] M. A. Lill and V. Helms. Molecular dynamics simulation of proton transport with quantum mechanically derived proton hopping rates (Q-HOP MD). J. Chem. Phys., 115:7993, 2001.
- [LH01b] M. A. Lill and V. Helms. Reaction rates for proton transfer over small barriers and connection to transition state theory. J. Chem. Phys., 115:7985, 2001.
- [Mar06] D. Marx. Proton Transfer 200 Years after Grotthus: Insights from Ab Initio Simulations. ChemPhysChem, 7(9):1848, 2006.
- [SW04] C. N. Schutz and A. Warshel. Analyzing Free Energy Relationships for Proton Translocations in Enzymes: Carbonic Anhydrase Revisited. J. Phys. Chem. B, 108:2066–2075, 2004.
- [TH03a] S. Taraphder and G. Hummer. Dynamic Proton Transfer Pathways in Proteins: Role of Sidechain Conformational Fluctuations. Physica A, 318:293–301, 2003.
- [TH03b] S. Taraphder and G. Hummer. Protein Side-Chain Motion and Hydration in Proton-Transfer Pathways. Results for Cytochrome P450cam. J.AM.CHEM.SOC, 125:3931–3940, 2003.