Publication
Eyrisch, S. and Helms, V. (2007) Journal of Medicinal Chemistry, vol. 50, p. 3457-3464, doi:10.1021/jm070095g. Transient pockets on protein surfaces involved in protein-protein interaction.
Motivation
The identification of ligand binding pockets is of great importance in structure-based drug design. Although many algorithms (e.g. PASS [1], LigSite [2], SURFNET [3]) have been presented that computationally detect cavities on protein surfaces they are designed to predict binding sites in a single protein structures. Thus, they only give a static description of the pocketome. In cases where the ligand binding pocket is unknown and no suitable pocket could be identified in a single (often unbound) protein conformation, it is recommendable to analyze all pockets accessible in a conformational ensemble of this protein [4,5]. Such a conformational ensemble may e.g. be generated using tCONCOORD [6] or be extracted from molecular dynamics simulations. The resulting dynamic description of the pocketome then contains transient pockets that are only open in some of the protein conformations but may represent putative ligand binding pockets.
Implementation
In EPOSBP, the PASS algorithm was re-implemented in C++ using the BALL library [7]. All parameters (minimum separation between two pockets, probe radii, etc.) are read in from a parameter file and thus can be adjusted. Further, we included the calculation of the pocket properties volume, polarity, and depth. The program writes two output files per detected pocket in PDB format: the “patch file” (the negative image of the pocket formed by the probes used by the algorithm for detecting the pocket) and the file containing the “pocket lining atoms” (all protein atoms found within a distance of 5 Å from the pocket probes).
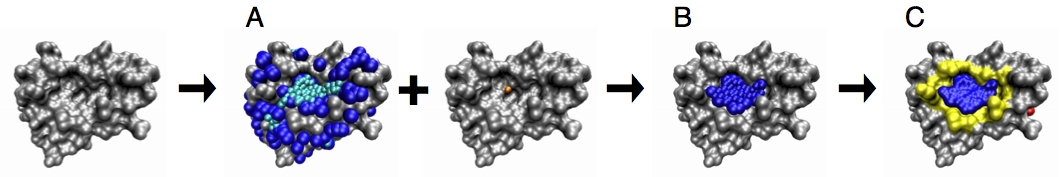
Calculating Ensembles of Pockets
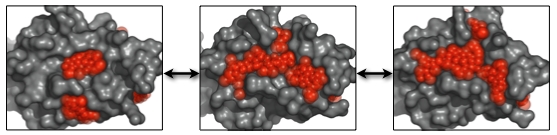
We extended the PASS algorithm for enabling the identification of the dynamic pocketome available in a conformational ensemble. To this end, cavities are detected in each input protein conformation and clustered using the pocket lining atoms (PLAs). All pockets assigned to the same cluster are then defined to be states of the same dynamic pocket and labeled by the same pocket identifier (PID). Finally, the dynamic properties of all pockets are analyzed, including the calculation of their relative frequency, and their average, minimum and maximum pocket properties, namely volume, polarity, and depth. The results are written to an output file.
Two sets of dynamic pockets resulting from different conformational ensembles can be efficiently compared by calculating the similarities of the so-called “subpockets”. A subpocket of a dynamic pocket is defined as the set of PLAs that appeared in a given percentage of its pocket states. The similarity of subpockets can also be calculated using EPOSBP.
Furthermore, the overlap of a pocket with a known ligand can be calculated. Especially when studying the dynamics of a known ligand binding pocket, it may be interesting to approximate the degree to which the native binding pocket is open in the set of conformations.
References
- Brady, G.P. and Stouten, P.F.W. (2000) Fast prediction and visualization of protein binding pockets with PASS. J. Comput. Aided Mol. Des., 14, 383-401.
- Hendlich, M. et al. (1997) LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model., 15, 359-363.
- Laskowski, R.A. (1995) SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph., 13, 323-330.
- Eyrisch, S. and Helms, V. (2007) Transient pockets on protein surfaces involved in protein-protein interaction. J. Med. Chem., 50, 3457-6364.
- Eyrisch, S. and Helms, V. (2008) What induces pocket openings on protein surface patches involved in protein-protein interactions? J. Comput. Aided Mol. Des. (accepted).
- Seelinger, D., Haas, J., de Groot, B.L. (2007) Geometry-based sampling of conformational transitions in proteins. Structure, 15, 1482-1492.
- Kohlbacher, O. and Lenhof, H.P. (2000) BALL – Rapid Software Prototyping in Computational Molecular Biology. Bioinformatics, 16, 815–824.